
Protein design facts for kids
Protein design is like being an architect for tiny building blocks inside living things. It's about creating new protein molecules with specific jobs, behaviors, or purposes. Scientists do this to understand how proteins work and to make new tools for medicine or industry.
Proteins can be designed in two main ways. One way is to build them completely from scratch. This is called de novo design. The other way is to change an existing protein's structure or sequence. This is known as protein redesign.
When scientists design proteins, they predict which parts (called amino acids) will make a protein fold into a certain shape. They then test these predictions in the lab. They might use methods like making new peptides or changing specific parts of a gene.
Scientists started designing proteins in the mid-1970s. Today, we have much better tools and understanding. This has led to many successful designs of new proteins. Some of these can even work inside cell membranes.
What is Protein Design?
The main goal in protein design is to figure out which sequence of amino acids will fold into a specific protein shape. Imagine you have a long chain of beads. How you arrange those beads decides the final 3D shape of the chain.
There are a huge number of possible protein sequences. But only a few of them will fold correctly and quickly into a stable shape. This stable shape is called the protein's "native state." Protein design is about finding these special sequences.
The native state is the most comfortable and stable shape for a protein. So, protein design is like searching for sequences that naturally prefer to be in a chosen shape. It's the opposite of protein structure prediction. In prediction, you know the sequence and try to guess the shape. In design, you know the shape and try to find the sequence. This is why it's sometimes called inverse folding.
Protein design is a puzzle where scientists use scoring rules to pick the best sequence. This sequence will then fold into the desired structure.
A Brief History of Protein Design
The first proteins were designed in the 1970s and 1980s. Back then, scientists designed them by hand. They looked at other known proteins and thought about the best amino acids to use. They also considered the desired shape.
Bernd Gutte is known for designing some of the first proteins. He created a simpler version of a known enzyme. He also designed structures with specific shapes like beta-sheets and alpha-helices. Later, other scientists designed stretchy, fiber-like proteins.
In the 1990s, powerful computers became available. This changed protein design a lot. Scientists developed computer tools that could help design proteins. These tools used libraries of amino acid shapes and special rules (called force fields).
Over the last 30 years, protein design has seen great success. In 1997, Stephen Mayo and his team designed the first protein completely from scratch. This was a huge step! Soon after, in 1999, Peter S. Kim's team designed new coiled-coil structures.
In 2003, David Baker's lab designed a full protein with a shape never seen before in nature. Later, in 2008, his group even designed new enzymes using computers. These enzymes could help with different chemical reactions.
In 2010, scientists used a designed protein to find a powerful antibody. This antibody can fight the HIV virus. Because of these and other successes, protein design is now a very important tool. Scientists hope to use it to create new proteins for medicine and engineering.
Images for kids
-

The Top7 protein was one of the first proteins designed for a fold that had never been seen before in nature
-
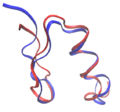
FSD-1 (shown in blue, PDB id: 1FSV) was the first de novo computational design of a full protein. The target fold was that of the zinc finger in residues 33–60 of the structure of protein Zif268 (shown in red, PDB id: 1ZAA). The designed sequence had very little sequence identity with any known protein sequence.
-

Common protein design programs use rotamer libraries to simplify the conformational space of protein side chains. This animation loops through all the rotamers of the isoleucine amino acid based on the Penultimate Rotamer Library (total of 7 rotamers).
-

Comparison of various potential energy functions. The most accurate energy are those that use quantum mechanical calculations, but these are too slow for protein design. On the other extreme, heuristic energy functions are based on statistical terms and are very fast. In the middle are molecular mechanics energy functions that are physically based but are not as computationally expensive as quantum mechanical simulations.
-
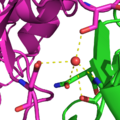
Water-mediated hydrogen bonds play a key role in protein–protein binding. One such interaction is shown between residues D457, S365 in the heavy chain of the HIV-broadly-neutralizing antibody VRC01 (green) and residues N58 and Y59 in the HIV envelope protein GP120 (purple).
-
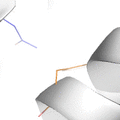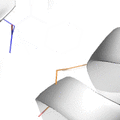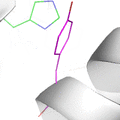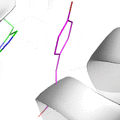
This animation illustrates the complexity of a protein design search, which typically compares all the rotamer-conformations from all possible mutations at all residues. In this example, the residues Phe36 and His 106 are allowed to mutate to, respectively, the amino acids Tyr and Asn. Phe and Tyr have 4 rotamers each in the rotamer library, while Asn and His have 7 and 8 rotamers, respectively, in the rotamer library (from the Richardson's penultimate rotamer library). The animation loops through all (4 + 4) x (7 + 8) = 120 possibilities. The structure shown is that of myoglobin, PDB id: 1mbn.





See also
- Molecular design software
- Protein engineering
- Protein structure prediction software
- Comparison of software for molecular mechanics modeling
