
Spinal muscular atrophy facts for kids

Spinal muscular atrophy (SMA) is a serious genetic condition that makes nerves and muscles stop working properly. Babies with SMA often become very weak. They might have trouble breathing because their lungs and breathing muscles are affected. Adults who get SMA slowly lose the ability to move their bodies. This muscle weakness usually gets worse as a person gets older.
SMA is passed down from parents. Both parents might carry a faulty gene that causes SMA, even if they don't have the disease themselves. About 1 in 25 to 1 in 50 people carry this faulty gene, depending on the country.
Spinal muscular atrophy is the most common genetic disease that causes deaths in babies. There are medicines that target the genetic cause of SMA, like nusinersen, risdiplam, and a gene therapy called onasemnogene abeparvovec. Other helpful care includes physical therapy, occupational therapy, help with breathing, good nutrition, bone and joint care, and support for moving around.
Contents
What are the Different Types of SMA?
SMA comes in different types, from very severe to milder forms. The type depends on when symptoms start and how serious they are.
Type 0: The Most Severe Form
This type can be seen even before a baby is born. It is the rarest and most serious form of SMA. Babies with Type 0 SMA often move less inside the womb. They might be born with joints that are bent or stiff. They have very weak muscles at birth. Their breathing muscles are also very weak. Most babies with Type 0 SMA do not live past their first year because of breathing problems. Some also have heart problems from birth.
Type I: Werdnig-Hoffmann Disease
This is the most common type of SMA. It is a severe form that shows up at birth or within the first few months. Children with Type I SMA cannot control their head movements. They may struggle to sit up without help. Many also have difficulty swallowing, which makes feeding hard. Breathing problems are common because their breathing muscles are weak. Most children with this type do not live past early childhood due to breathing issues.
Type II: Dubowitz Disease
Muscle weakness in Type II SMA usually starts when children are between 6 and 12 months old. They often cannot sit without support. They also usually cannot stand or walk without help. Other signs include shaky fingers and a curved spine (scoliosis). Weak breathing muscles can also be dangerous. Many people with Type II SMA live into their twenties or thirties.
Type III: Kugelberg-Welander Disease
This type causes muscle weakness in mid-childhood. Many children with Type III SMA can stand and walk on their own at first. But as they get older, walking and climbing stairs become much harder. Most people with this condition will need a wheelchair later in life. Unlike other types, people with Type III SMA usually have a normal life span.
Type IV: Adult-Onset SMA
This type is rare and usually appears in early adulthood. People with Type IV SMA often have mild to moderate muscle weakness. They might also have tremors (shaking) and very mild breathing problems.
How is SMA Managed and Treated?
Managing SMA depends on how severe it is and which type a person has. People with very severe forms (like Type 0 or I) need help right away. Those with milder forms (like Type IV) might not need certain care until much later in life. Since each person is different, their specific care plan can also be different.
Medications for SMA
There are several medicines approved to treat spinal muscular atrophy:
- Nusinersen (called Spinraza) is given directly into the fluid around the central nervous system. It helps babies with SMA live longer and move better. It was approved in the US in 2016 and in Europe in 2017.
- Onasemnogene abeparvovec (called Zolgensma) is a gene therapy. It was first approved in the US in May 2019 for children under 24 months. It is given through an IV. Other countries have also approved it.
- Risdiplam (called Evrysdi) is a liquid medicine taken by mouth. It was approved in the US in August 2020 and is now approved in many countries.
Breathing Support
Breathing problems are the most common and serious issues in SMA Types 0, I, and II. They are the main cause of death. Type III SMA can also have breathing problems, but it's less common. These problems happen because the muscles between the ribs become weak. The diaphragm, a main breathing muscle, is less affected. Once these muscles weaken, they don't fully recover. This makes breathing and coughing harder. People might not get enough oxygen, especially when they sleep.
It also becomes harder to clear mucus from the airways. Swallowing muscles can be affected, leading to food or liquid going into the lungs. This increases the risk of infections like pneumonia. To help clear airways, people might use chest physical therapy or special cough assistance devices. For breathing, Non-invasive ventilation (like BiPAP) is often used. Sometimes, a tracheostomy (a tube in the windpipe) is needed in severe cases. Both methods help people live longer, but a tracheostomy can make it hard to speak.
Nutrition and Eating
The more severe the SMA type, the more likely a person is to have problems with nutrition. These can include difficulty eating, opening the jaw, chewing, and swallowing. Such difficulties can lead to not getting enough food or too much, not growing well, or food going into the lungs. Other issues, especially for those who can't walk, include food moving too slowly through the stomach, reflux, constipation, vomiting, and bloating.
Sometimes, people with severe SMA (Type I and some Type II) might need a feeding tube or a gastrostomy (a tube directly into the stomach). It's also suggested that people with SMA, especially severe forms, eat less fat and eat more often to avoid long periods without food. Choosing softer foods can help prevent choking. During an illness, nutritional problems can get worse or appear for the first time.
Orthopaedic Care for Bones and Joints
Weak muscles in SMA can lead to bone and joint problems. These include stiff joints, hip dislocations, curved spines, weak bones, a higher risk of fractures, and pain. Weak muscles that normally support the spine can cause it to curve (kyphosis or scoliosis). Sometimes, Spine fusion surgery is done for people with SMA Type I or II around ages 8-10. This helps relieve pressure on the lungs from a curved spine.
For people who can't move much, good posture and position in wheelchairs are important. Range of motion exercises and bone-strengthening activities can help prevent problems. Many people with SMA also benefit a lot from different types of physiotherapy and occupational therapy.
Special devices called orthotics can support the body and help with walking. For example, AFOs (ankle foot orthoses) support the foot and help with walking. TLSOs (thoracic lumbar sacral orthoses) support the upper body. Assistive technologies can also help with movement and daily activities, greatly improving life quality.
Other Important Aspects
While the heart is not usually a main concern, some studies suggest a link between SMA and certain heart conditions.
Children with SMA usually act like other children their age. Their cognitive development (how they learn and think) can even be a bit faster. Some parts of their intelligence might be above average. Despite their physical challenges, many people with SMA report being very happy with their lives.
What is the Outlook for People with SMA?
Without specific medical treatment, people with SMA tend to get weaker over time. However, with good and early supportive care for breathing and nutrition, survival has improved for those with severe SMA.
If not treated, most children with SMA Type 0 and I do not live past age 4. Breathing problems are the main cause of death. With proper care, some milder cases of SMA Type I (about 10% of all Type I cases) can live into adulthood. More research is needed on long-term survival for Type I, but modern breathing support seems to have reduced deaths.
For untreated SMA Type II, the disease progresses more slowly. Life expectancy is less than for healthy people. Death before age 20 is common, but many people with SMA Type II live to become parents and grandparents. SMA Type III usually has a normal or near-normal life expectancy if good care is followed. Type IV, which starts in adulthood, usually only causes movement problems and does not affect how long a person lives.
Images for kids
-
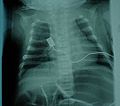
This X-ray shows a bell-shaped torso. This can happen when the muscles between the ribs are weak and a person uses their stomach muscles to breathe. A bell-shaped torso is not only seen in people with SMA.

See also
 In Spanish: Atrofia muscular espinal para niños
In Spanish: Atrofia muscular espinal para niños
