
Elias James Corey facts for kids
Quick facts for kids
E.J. Corey
|
|
|---|---|

Corey in 2007
|
|
| Born |
Elias James Corey
July 12, 1928 Methuen, Massachusetts, U.S.
|
| Alma mater | Massachusetts Institute of Technology (BS, PhD) |
| Known for |
|
| Awards |
|
| Scientific career | |
| Fields | Organic chemistry |
| Institutions | University of Illinois at Urbana–Champaign Harvard University |
| Thesis | The synthesis of N,N-diacylamino acids and analogs of penicillin (1951) |
| Doctoral advisor | John C. Sheehan |
| Notable students |
|
Elias James Corey (born July 12, 1928), also known as E.J. Corey, is a famous American organic chemist. He won the Nobel Prize in Chemistry in 1990 for creating new ways to build complex molecules. His most famous idea is called retrosynthetic analysis, which is like planning a project by starting at the end and working backward.
Contents
Early Life and Education
E.J. Corey was born in Methuen, Massachusetts. His parents were Christian immigrants from Lebanon. His father died when he was only 18 months old. To honor him, his mother changed his name from William to Elias.
Growing up during the Great Depression, his family faced hard times. Corey was an independent kid who loved sports like baseball and football. He went to a Catholic elementary school and later Lawrence High School.
At just 16, he started at the Massachusetts Institute of Technology (MIT). He first planned to study engineering but fell in love with chemistry after his first class. He earned his bachelor's degree in 1948 and his Ph.D. in chemistry in 1951.
A Career in Chemistry
After graduating, Corey became a professor at the University of Illinois at Urbana–Champaign. By age 27, he was already a full professor. In 1959, he moved to Harvard University, where he has worked for most of his career.
Corey chose to study organic chemistry because he found it beautiful and very important for human health. For over 50 years, he also worked as an advisor for the drug company Pfizer.
Throughout his career, Corey has won many awards. These include the National Medal of Science in 1988 and the Nobel Prize in Chemistry in 1990.
Major Discoveries in Chemistry
E.J. Corey is famous for developing new tools and methods that chemists use to make important molecules, like medicines.
New Chemical Tools
Corey and his team created several new chemical substances, called reagents, that help with chemical reactions.
- PCC (pyridinium chlorochromate): Also known as the Corey-Suggs reagent, this is a special chemical that helps turn alcohols into other useful compounds called aldehydes and ketones. It is a stable, easy-to-use solid.
- Protecting Groups: When chemists build a large molecule, they sometimes need to protect certain parts of it from changing during a reaction. Corey developed popular "protecting groups" like TBS and TIPS. These act like temporary shields for parts of a molecule, which can be removed later. This allows chemists to build very complex natural products.
New Chemical Methods
Corey also invented many new types of chemical reactions, which are like recipes for making molecules. Many of these are now named after him.
- Corey-Itsuno Reduction: This is a clever way to turn one type of molecule (a ketone) into a specific type of alcohol. It's special because it creates a "chiral" molecule, meaning it produces a specific "left-handed" or "right-handed" version of the product.
- Corey-Fuchs Reaction: This method is used to create a type of molecule called a terminal alkyne from an aldehyde. It's a reliable way to add a carbon atom and create a special chemical bond.
- Corey–Kim Oxidation: This is another way to turn alcohols into aldehydes and ketones. It's a good alternative to other methods because it uses less toxic chemicals.
- Corey-Winter Olefination: This reaction turns a molecule called a 1,2-diol into an alkene. It is very precise and creates a specific shape of the final molecule.
- Corey-Nicolaou Macrolactonization: This was the first good method for creating very large, ring-shaped molecules called macrolactones. These are found in many important natural products and medicines.
Building Complex Molecules
Using his new methods, E.J. Corey and his team were able to build many complex molecules found in nature. This is called total synthesis. By 2010, his group had successfully made at least 265 of these natural compounds.
One of his most famous achievements was the synthesis of prostaglandins in 1969. Prostaglandins are hormone-like substances in the body that have many important jobs. Making them in a lab was very difficult because of their complicated structure.
Corey's plan for making prostaglandins used his idea of retrosynthetic analysis. He started with the final molecule and worked backward to figure out the simplest starting materials. This smart planning made a very hard problem much easier to solve.
The Woodward–Hoffmann Rules Controversy
In 2004, Corey said that he gave the main idea for the famous Woodward–Hoffmann rules to another chemist, Robert Burns Woodward, in 1964. These rules are a very important theory in chemistry.
Corey stated that he suggested the idea to Woodward, who then presented it as his own. Corey said he kept quiet about it for many years because he did not want to cause problems for Harvard, where they both worked.
Another scientist, Roald Hoffmann, who shared the Nobel Prize for this work with Woodward, disagreed with Corey's story. Woodward had passed away in 1979 and could not give his side of the story. This disagreement remains a topic of discussion among chemists.
Legacy and Honors
E.J. Corey is one of the most influential chemists of the 20th century. His work has changed how scientists approach making molecules. Many of his students have also become famous chemists.
He has received over 40 major awards and 19 honorary degrees from universities around the world. In 2013, the E.J. Corey Institute of Biomedical Research (CIBR) opened in China, named in his honor.
Images for kids
-

A diagram showing how the PCC reagent works.
-
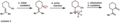
This diagram shows how PCC can be used to form ring-shaped molecules.
-
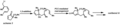
An example of a complex rearrangement reaction using PCC.
-
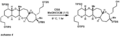
How a "TBS" protecting group is removed from a molecule.
-
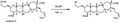
A diagram showing the removal of a "TIPS" protecting group.
-
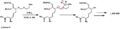
A diagram showing how a "MEM" protecting group is removed.
-
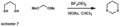
How a dithiane is formed from a carbonyl group.
-
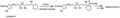
The Corey-Seebach reaction, which uses a dithiane to build a new molecule.
-
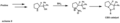
The creation of the CBS catalyst, a tool for making specific "handed" molecules.
-
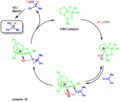
How the CBS catalyst works to guide a reaction.
-
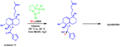
Using the CBS reduction to help build a natural product called dysidiolide.
-
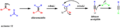
The steps of the Corey-Fuchs reaction for making an alkyne.
-
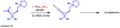
An example of the Corey-Fuchs reaction used in making a natural product.
-
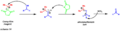
The mechanism of the Corey-Kim oxidation.
-

Using the Corey-Kim oxidation to change one part of a complex molecule.
-

The Corey-Winter reaction used to make a natural product called (+)-Boesenoxide.
-

A model of the transition state for an enantioselective Diels-Alder reaction.
-
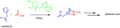
Using the Diels-Alder reaction to form a six-membered ring in a total synthesis.
-
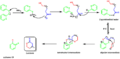
The steps of the Corey-Nicolaou macrolactonization for making large rings.
-
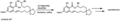
Using macrolactonization to make the natural product zearalenone.
-
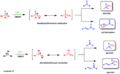
The Johnson-Corey-Chaykovsky reaction can make different products depending on the reagent used.
-

An example of the Corey-Chaykovsky reaction used in the synthesis of taxol.
-
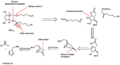
Corey's "retrosynthesis" plan for making prostaglandins.
-

Using the CBS reduction to create a key part of the prostaglandin molecule with high precision.






See also
 In Spanish: Elias James Corey para niños
In Spanish: Elias James Corey para niños
