
Sigmatropic reaction facts for kids
A sigmatropic reaction is a special type of chemical reaction in organic chemistry. Think of it like a puzzle where parts of a molecule rearrange themselves! In these reactions, a single bond (called a sigma bond) moves to a new spot within the same molecule. No new atoms are added, and no atoms leave.
The word "sigmatropic" comes from "sigma," which is the name for a single carbon-carbon bond, and the Greek word tropos, meaning "turn" or "change." So, it's about a sigma bond turning or shifting.
These reactions usually happen without a catalyst (something that speeds up a reaction). They are also called rearrangement reactions because the atoms and bonds simply shift around inside one molecule. Some well-known sigmatropic reactions include the Cope rearrangement, Claisen rearrangement, and Fischer indole synthesis.
Contents
How We Name Sigmatropic Shifts
Chemists use a special way to describe sigmatropic shifts, called [i,j] notation. This helps us understand how the atoms move.
Understanding the [i,j] Numbers
To find the [i,j] numbers for a sigmatropic shift, you first find the bond that breaks. You count the atoms from each end of this breaking bond to the atoms that form the new bond.
- Start by numbering the atoms of the bond that is breaking as "1" on both sides.
- Then, count along the molecule's "backbone" (the main chain of atoms) in each direction until you reach the atoms that will form the new sigma bond.
- The numbers you get for these new bond-forming atoms are placed in square brackets, separated by a comma, like [1,5] or [3,3]. This is the "order" of the shift.
For example, if a hydrogen atom moves, we count all the atoms involved in the chain, not just the closest ones. This helps us get the correct [i,j] number.


Suprafacial and Antarafacial Shifts
When a group of atoms moves in a sigmatropic reaction, it can move in two main ways:
- Suprafacial: The moving group stays on the same side or "face" of the molecule's pi system (a special cloud of electrons).
- Antarafacial: The moving group goes to the opposite side or "face" of the molecule's pi system.
Think of it like flipping a pancake (antarafacial) versus sliding it (suprafacial). For molecules that are small or in rings, it's often hard for them to do an antarafacial shift because of how they are shaped.

Types of Sigmatropic Rearrangements
[1,3] Shifts
In a [1,3] shift, a group of atoms moves three atoms away from its original spot.
Thermal Hydride Shifts
A "hydride" is a hydrogen atom with an extra electron. In a thermal [1,3] hydride shift, a hydride moves. The rules of chemistry say this shift should happen in an antarafacial way (moving to the opposite face). However, this is usually impossible for molecules to do because of their shape. This is why some common molecules don't rearrange easily without help from an acid or base.

Thermal Alkyl Shifts
"Alkyl" groups are chains of carbon and hydrogen atoms. Like hydride shifts, thermal [1,3] alkyl shifts should also happen antarafacially. But alkyl groups can sometimes flip their shape to make a new bond, allowing a suprafacial shift. These reactions are still not very common unless the molecule is in a ring shape.

Photochemical [1,3] Shifts
"Photochemical" means the reaction is started by light. Photochemical [1,3] shifts are expected to be suprafacial. However, many of them don't happen in a single, smooth step. Instead, they go through a "diradical" stage, which is a bit different.
[1,5] Shifts
A [1,5] shift involves a group moving five atoms down a pi system.
- Hydrogen shifts: Hydrogen atoms can shift in both open-chain and ring-shaped molecules when heated to high temperatures (around 200°C). These shifts are expected to be suprafacial.

- Alkyl shifts: Unlike hydrogen, alkyl groups don't easily undergo [1,5] shifts in open-chain molecules. They usually need very high temperatures. However, in some ring molecules like cyclohexadienes, alkyl shifts can happen more easily. This is because the ring first opens up, then a [1,7] shift happens, and finally the ring closes again.

[1,7] Shifts
[1,7] sigmatropic shifts are predicted to happen in an antarafacial way (moving to the opposite face). An example is seen in how lumisterol changes into vitamin D2. After a ring opens, a hydrogen atom from a methyl group shifts.

[3,3] Shifts
[3,3] sigmatropic shifts are very well-known and studied. They involve six electrons and are predicted to happen suprafacially.
Claisen Rearrangement
The Claisen rearrangement was discovered in 1912. It's a very useful reaction for making new carbon-carbon bonds. When a molecule called an allyl vinyl ether is heated, it rearranges to form a special kind of molecule called a γ,δ-unsaturated carbonyl. This reaction is special because it creates a carbonyl group, which means it usually can't go backward.

Aromatic Claisen Rearrangement
This is a version of the Claisen rearrangement that happens with molecules that have a benzene ring (a special type of ring). An allyl phenyl ether rearranges to form a phenol. If both "ortho" positions (spots next to where the original group was attached) on the benzene ring are blocked, a second rearrangement can happen, leading to a "para-Claisen rearrangement."


Cope Rearrangement
The Cope rearrangement was developed by Arthur C. Cope. It's a [3,3] sigmatropic rearrangement that happens with molecules called 1,5-dienes. For example, if you heat 3,4-dimethyl-1,5-hexadiene to 300°C, it changes into 2,6-octadiene.

Oxy-Cope Rearrangement
The Oxy-Cope rearrangement is a variation where a hydroxyl group (an -OH group) is added to the molecule. After the rearrangement, this leads to the formation of an enal or enone.

Carroll Rearrangement
The Carroll rearrangement is another rearrangement reaction. It changes a special kind of ester (a β-keto allyl ester) into an α-allyl-β-ketocarboxylic acid. This reaction is often followed by another step where a carbon dioxide molecule is removed, resulting in a γ,δ-allylketone. It's like a special version of the Claisen rearrangement that also removes a part of the molecule.

Fischer Indole Synthesis
The Fischer indole synthesis is a chemical reaction that makes a molecule called indole. Indole is an "aromatic heterocycle," which means it's a ring-shaped molecule with special stability and contains atoms other than just carbon in its ring. This reaction uses a phenylhydrazine and an aldehyde or ketone, usually with an acid to help it along. Hermann Emil Fischer discovered it in 1883.

The type of acid used is important. Strong acids like HCl or H2SO4 work well. Other acids like boron trifluoride or zinc chloride can also be used.
[5,5] Shifts
Similar to [3,3] shifts, [5,5] sigmatropic shifts are also predicted to happen suprafacially. These reactions are less common than [3,3] shifts, mainly because the molecules that can undergo [5,5] shifts are not as common.

Walk Rearrangements
A "walk rearrangement" happens when a group of atoms that is part of a three-membered ring moves within a larger bicyclic molecule (a molecule with two rings). It's like a small part of the molecule "walking" to a new position.
An example is the rearrangement of tropilidenes. When heated, their pi-system closes into a ring, then a [1,5] alkyl shift happens, and finally the ring opens again.

Images for kids
-
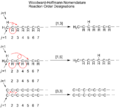
Detailed Woodward-Hoffmann nomenclature.
-
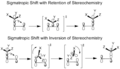
Stereochemistry: retention vs. inversion.


Related pages
- 2,3-sigmatropic rearrangement
- NIH shift
- Frontier Molecular Orbital Theory
- Woodward–Hoffmann rules
See also
 In Spanish: Reacción sigmatrópica para niños
In Spanish: Reacción sigmatrópica para niños
