
Sickle cell disease facts for kids
Quick facts for kids Sickle cell disease |
|
|---|---|
| Synonyms | Sickle cell disorder |
 |
|
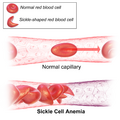
| Figure (A) shows normal red blood cells flowing freely through a blood vessel. The inset shows a cross-section of a normal red blood cell with normal haemoglobin. Figure (B) shows abnormal, sickled red blood cells sticking at the branching point in a blood vessel. The inset image shows a cross-section of a sickle cell with long polymerized sickle haemoglobin (HbS) strands stretching and distorting the cell shape to look like a crescent moon. | |
| Symptoms | Attacks of pain, anemia, swelling in the hands and feet, bacterial infections, stroke |
| Complications | Chronic pain, stroke, aseptic bone necrosis, gallstones, leg ulcers, pulmonary hypertension, vision problems, kidney problems |
| Usual onset | 5–6 months of age |
| Causes | Genetic, Homozygous mutation in the hemoglobin S gene. |
| Diagnostic method | Blood test |
| Treatment | Vaccination, antibiotics, high fluid intake, folic acid supplementation, pain medication, blood transfusions |
| Prognosis | Life expectancy 40–60 years (developed world) |
| Frequency | 4.4 million (2015) |
| Deaths | 114,800 (2015) |
Sickle cell disease (SCD) is a group of blood disorders that people usually get from their parents. The most common type is called sickle cell anaemia. It affects a protein in your red blood cells called haemoglobin, which carries oxygen.
Because of this problem, red blood cells can become stiff and shaped like a crescent moon or "sickle" under certain conditions. These problems often start when a baby is around 5 or 6 months old. People with SCD can have different health issues, like sudden attacks of pain (called a sickle cell crisis), anemia (not enough healthy red blood cells), swelling in their hands and feet, bacterial infections, and even strokes. As people get older, they might also have long-lasting pain. In many developed countries, people with SCD live to be about 40 to 60 years old.
Sickle cell disease happens when a person gets two unusual copies of a special gene (called HBB) that helps make haemoglobin. They get one copy from their mom and one from their dad. There are different types of SCD, depending on the exact change in these genes. Things like changes in temperature, stress, not drinking enough water (dehydration), and being at high places (high altitude) can make symptoms worse. If someone has only one unusual copy of the gene, they usually don't have symptoms. They are called a carrier or someone with sickle cell trait.
Doctors can find out if someone has SCD with a blood test. Some countries even test all babies for the disease right after they are born. It's also possible to diagnose it during pregnancy.
Caring for people with sickle cell disease often includes preventing infections with vaccinations and antibiotics. It's also important to drink lots of fluids, take folic acid supplements, and use pain medication. Sometimes, people might need blood transfusions or a medicine called hydroxycarbamide. In rare cases, a bone marrow transplant can cure the disease.
In 2015, about 4.4 million people worldwide had sickle cell disease. Another 43 million people had sickle cell trait. Most cases (about 80%) are found in Sub-Saharan Africa. It's also seen in parts of India, Southern Europe, West Asia, North Africa, and among people of African origin living in other parts of the world. In 2015, about 114,800 deaths were caused by SCD. An American doctor named James B. Herrick first described this condition in 1910. Later, in 1949, scientists figured out how it was passed down through families. In 1954, it was discovered that having sickle cell trait can protect against malaria.
Understanding Sickle Cell Disease



Signs of sickle cell disease usually appear when children are very young. How severe the symptoms are can be different for each person. SCD can lead to various health problems, both sudden (acute) and long-lasting (chronic). Some of these problems can be very serious.
Sickle Cell Crisis: Pain Attacks
The term "sickle cell crisis" describes different sudden problems that happen to people with SCD. These can cause anemia and pain. There are several types of crises, like the vaso-occlusive crisis (where blood flow is blocked), aplastic crisis, and others. Most sickle cell crises last about five to seven days. Sometimes, infections, not drinking enough water, or changes in body chemistry can trigger a crisis. However, often there's no clear reason why a crisis starts.
Where Sickle Cell Disease is Common
Sickle cell disease is most common in warm, tropical areas. This includes places like sub-Saharan Africa, parts of India, and the Middle East. In recent years, many people have moved from these areas to countries in Europe. Because of this, sickle cell disease is now more common in some European countries than other genetic conditions like haemophilia or cystic fibrosis. In 2015, about 114,800 people died from SCD.
SCD is more common among people whose ancestors lived in tropical and subtropical regions where malaria was widespread. In these areas, having one copy of the sickle cell gene (sickle cell trait) can be helpful. People with sickle cell trait often have less severe symptoms if they get malaria.
This condition is inherited in a specific way. It means that both copies of the gene in each cell must have a change (mutation). The parents each carry one copy of the changed gene. However, they usually do not show any signs or symptoms of the condition themselves.
New Discoveries and Treatments
Umbilical Cord Blood Transplants
A umbilical cord blood transplant can potentially cure sickle cell disease. However, it's hard to find a suitable donor for everyone; only about 10% of people find one. Also, about 7% of people who have this procedure might die from it. Another risk is something called graft versus host disease, where the new cells attack the patient's body.
Gene Therapy: Fixing the Genes
Diseases like sickle cell disease are good candidates for gene therapy. This is because a healthy copy of the gene can potentially fix the problem in the cells. However, the full risks and benefits of gene therapy for SCD are still being studied.
In 2001, scientists successfully treated sickle cell disease in mice using gene therapy. They used a special virus to help the mice produce fetal haemoglobin (HbF). Humans normally stop making HbF shortly after birth. In people, a medicine called hydroxyurea can temporarily help by increasing HbF production. The researchers showed that gene therapy could be a more lasting way to boost HbF.
Human trials for gene therapy for SCD began in 2014. These trials are checking if a new method using modified bone marrow is safe for adults with severe SCD. As of 2020, there haven't been large, controlled studies yet. The first person treated was reported in March 2017, and a few more people have been treated since then.
New tools like CRISPR/Cas9, which can edit genes, have been used to correct the gene change that causes SCD. This is done in stem cells taken from a person with the condition. In July 2019, the CRISPR tool was used to edit bone marrow cells from a person with SCD. This helped boost fetal haemoglobin by turning off a specific gene called BCL11A. Some researchers have also thought about the ethical questions of using CRISPR for SCD. This is because of past unfair treatment of the African American community by the medical field.
In 2017, twelve clinical trials were looking at gene therapy for sickle cell anaemia. Four of these trials aimed to replace the changed HBB gene with a healthy one. Three trials used a medicine called Mozobil to see if increasing stem cells could help with gene therapy. One trial studied bone marrow samples from patients with SCD. Another trial experimented with using umbilical cord blood from babies, both with and without SCD, to develop gene therapy.
In November 2023, the British Medical Journal reported exciting news. A gene treatment using the CRISPR gene editing tool was approved in the UK. This treatment is for both sickle cell disease and another blood disorder called β thalassaemia.
Stem Cell Transplants
There isn't strong medical proof yet to fully understand the risks and benefits of using hematopoietic stem cell transplants to treat people with sickle cell disease. More research is needed to be sure.
Images for kids
-

Figure (A) shows normal red blood cells flowing freely through a blood vessel. The inset shows a cross-section of a normal red blood cell with normal haemoglobin. Figure (B) shows abnormal, sickled red blood cells sticking at the branching point in a blood vessel. The inset image shows a cross-section of a sickle cell with long polymerized sickle haemoglobin (HbS) strands stretching and distorting the cell shape to look like a crescent moon.
-
Sickle cell anaemia
-
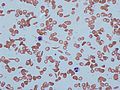
Sickle cells in human blood - both normal red blood cells and sickle-shaped cells are present.
-
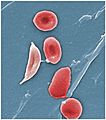
Normal blood cells next to a sickle blood cell, coloured scanning electron microscope image
