
Ehlers–Danlos syndromes facts for kids
Quick facts for kids Ehlers–Danlos syndromes (EDS) |
|
|---|---|
.png) |
|
| Individual with EDS displaying skin hyperelasticity | |
| Pronunciation | |
| Symptoms | Overly flexible joints, stretchy skin, abnormal scar formation |
| Complications | Aortic dissection, joint dislocations, osteoarthritis |
| Usual onset | Childhood or teens depending on type. |
| Duration | Lifelong |
| Types | Hypermobile, classic, vascular, kyphoscoliosis, arthrochalasia, dermatosparaxis, brittle cornea syndrome, others |
| Causes | Genetic |
| Risk factors | Family history |
| Diagnostic method | Genetic testing, physical examination |
| Similar conditions | Marfan syndrome, cutis laxa syndrome, familial joint hypermobility syndrome, Loeys–Dietz syndrome, hypermobility spectrum disorder |
| Treatment | Supportive |
| Prognosis | Depends on specific disorder |
| Frequency | 1 in 5,000 |
Ehlers–Danlos syndromes (EDS) are a group of 13 different genetic disorders. These conditions affect the body's connective tissue, which is like the "glue" that holds everything together.
People with EDS often have very flexible joints, skin that stretches easily, and scars that form in unusual ways. These signs might show up when a baby is born or during early childhood. Sometimes, EDS can lead to serious problems like joint dislocations or early osteoarthritis.
EDS happens because of changes in more than 19 different genes. These changes are present from birth. The specific gene affected decides which type of EDS a person has. Some people get EDS from their parents, while for others, the gene change happens on its own. These gene changes usually affect a protein called collagen, which is important for strong connective tissue.
Doctors often diagnose EDS based on a person's symptoms. They can confirm it with genetic testing or a skin biopsy. There is no cure for EDS, but treatments can help manage the symptoms. Physical therapy and braces can help make muscles stronger and support joints. Some types of EDS allow people to live a normal life, but types that affect blood vessels can be more serious.
The hypermobile type of EDS (hEDS) is the most common, affecting about 1 in 5,000 people worldwide. Other types are much rarer. Doctors named the syndromes after two physicians, Edvard Ehlers and Henri-Alexandre Danlos, who described them around 1900.
Contents
- What are the Types of EDS?
- Hypermobile EDS (hEDS)
- Classical EDS (cEDS)
- Vascular EDS (vEDS)
- Kyphoscoliosis EDS (kEDS)
- Arthrochalasia EDS (aEDS)
- Dermatosparaxis EDS (dEDS)
- Brittle-Cornea Syndrome (BCS)
- Classical-Like EDS (clEDS)
- Spondylodysplastic EDS (spEDS)
- Musculocontractural EDS (mcEDS)
- Myopathic EDS (mEDS)
- Periodontal EDS (pEDS)
- Cardiac-Valvular EDS (cvEDS)
- Common Signs and Symptoms
- What Causes EDS?
- How is EDS Diagnosed?
- How is EDS Managed?
- What is the Outlook for People with EDS?
- How Common is EDS?
- History of EDS
- EDS in Society and Culture
- EDS in Animals
- See also
What are the Types of EDS?
In 2017, experts classified 13 different types of EDS. Each type has its own specific ways that doctors diagnose it. These types are grouped by how they affect the body's collagen and other important proteins.
Hypermobile EDS (hEDS)
Hypermobile EDS (hEDS) is mostly known for making joints very flexible. This means both large and small joints can move beyond their normal range. People with hEDS might have joints that often partly or fully dislocate. Their skin is usually soft, smooth, and bruises easily. They might also have ongoing muscle or bone pain.
This type affects the skin less than other forms of EDS. There isn't a specific genetic test for hEDS yet. Doctors diagnose it based on a person's physical signs, family history, and other health issues.
How Do Genes Play a Role in Hypermobile EDS?
Most types of EDS have specific gene changes that can be found with genetic testing. However, for hEDS, the exact gene cause is still unknown. Scientists are working hard to find it.
Researchers are studying the genes of many people with hEDS. They are looking for a common gene change. One lab has found a "very strong candidate gene" for hEDS. This discovery could lead to new treatments.
Scientists are also looking at genes involved in heart problems, like issues with the aorta and mitral valve. These heart problems are sometimes seen in people with EDS. For example, a gene called DZIP1 is being studied. Changes in this gene can affect how the body makes collagen, which could explain "floppy" heart valves.
Classical EDS (cEDS)
Classical EDS (cEDS) causes skin to be very stretchy, fragile, and bruise easily. Joints are also very flexible. People with cEDS might have small, soft bumps called molluscoid pseudotumors over pressure points. They might also have small cysts containing fat.
Wounds can be harder to close and heal slowly. Sometimes, babies with cEDS might be slow to develop motor skills or have weak muscles. This type of EDS is caused by changes in genes like COL5A2, COL5A1, and sometimes COL1A1.
Doctors can often make a likely diagnosis by carefully checking a person's mouth, skin, and bones. Genetic testing is the most reliable way to confirm cEDS. There is no cure, but gentle exercise and anti-inflammatory medicines can help with pain. Protecting the skin from injuries is also important.
Vascular EDS (vEDS)
Vascular EDS (vEDS) is a very serious type. It causes skin to be thin, see-through, and bruise very easily. Blood vessels and internal organs are also very fragile and can rupture (break open) easily. People with vEDS are often shorter than average and have thin scalp hair.
They might have specific facial features, like large eyes and a small chin. Joint flexibility is usually limited to fingers and toes. Other signs can include club foot, early varicose veins, and a collapsed lung. This type is mainly caused by changes in the COL3A1 gene.
Kyphoscoliosis EDS (kEDS)
Kyphoscoliosis EDS (kEDS) involves very weak muscles at birth and slow motor development. People with kEDS often have severe and progressive scoliosis (a curved spine) from birth. Their eyes might have fragile white parts (sclera) and unusually small corneas.
They can also bruise easily and have fragile arteries. Some people with kEDS have a "marfanoid habitus," meaning they are tall with long, slender fingers and limbs. This type is caused by changes in the PLOD1 or FKBP14 genes.
Arthrochalasia EDS (aEDS)
Arthrochalasia EDS (aEDS) is known for very severe joint flexibility and hip dislocations present at birth. Other common signs include fragile, stretchy skin that bruises easily, weak muscles, and a curved spine. This type is very rare, with only about 30 cases reported. It is caused by changes in the COL1A1 and COL1A2 genes.
Dermatosparaxis EDS (dEDS)
Dermatosparaxis EDS (dEDS) causes extremely fragile skin that leads to severe bruising and scarring. The skin can also be saggy, especially on the face. People with dEDS might have mild to serious joint flexibility and hernias. This type is extremely rare, with only about 11 cases reported. It is caused by changes in the ADAMTS2 gene.
Brittle-Cornea Syndrome (BCS)
Brittle-cornea syndrome (BCS) causes the cornea (the clear front part of the eye) to get progressively thinner. This can lead to severe nearsightedness and problems with the shape of the eye. People with BCS often have hearing loss and blue sclerae (the whites of their eyes). They also commonly have flexible joints and stretchy skin. This syndrome has two types, caused by changes in the ZNF469 or PRDM5 genes.
Classical-Like EDS (clEDS)
Classical-like EDS (clEDS) features very stretchy skin that feels velvety, but without the thin, "cigarette paper" scars seen in classical EDS. People have general joint flexibility, sometimes with repeated dislocations. They also bruise easily. This type is caused by changes in the TNXB gene.
Spondylodysplastic EDS (spEDS)
Spondylodysplastic EDS (spEDS) causes short stature that gets worse during childhood. People have weak muscles, which can range from severe at birth to mild later on. They might also have bowed limbs. This type can be caused by changes in the B4GALT7, B3GALT6, or SLC39A13 genes. Depending on the gene, people might have curved spines, thin fingers, weak bones, or eye problems.
Musculocontractural EDS (mcEDS)
Musculocontractural EDS (mcEDS) is known for tight joints (contractures) present at birth, especially in the hips, elbows, and feet (clubfoot). People also have distinct facial features that are noticeable early in life. Skin is stretchy, bruises easily, and can be fragile with thin scars. This type is caused by changes in the CHST14 or DSE genes.
Myopathic EDS (mEDS)
Myopathic EDS (mEDS) involves weak muscles and/or muscle loss that often gets better with age. People have tight joints in the knees, hips, and elbows, but flexible joints in the ankles, wrists, and hands. Doctors confirm this diagnosis with genetic testing for changes in the COL12A1 gene.
Periodontal EDS (pEDS)
Periodontal EDS (pEDS) is mainly characterized by severe, ongoing gum disease that starts early in childhood or teenage years. People also lack attached gum tissue and might have specific skin plaques on their shins. A family history of pEDS is also important for diagnosis. Genetic testing can find changes in the C1R or C1S genes.
Cardiac-Valvular EDS (cvEDS)
Cardiac-valvular EDS (cvEDS) is known for serious and worsening heart valve problems, especially affecting the aortic valve and mitral valve. People also have stretchy skin, thin scars, and bruise easily. Joint flexibility can be general or limited to small joints. This type is caused by changes in both copies of the COL1A2 gene.
Common Signs and Symptoms
EDS affects connective tissue throughout the body. This means symptoms can show up in many unexpected ways. The most common signs are seen in the joints, skin, and blood vessels. Symptoms can be very mild or life-threatening. Because there are many types of EDS, symptoms can be very different from person to person.
Musculoskeletal System
- Flexible Joints: Joints can be too flexible, making them unstable.
- Sprains and Dislocations: Joints are prone to sprains, partial dislocations, and full dislocations.
- Early Arthritis: People might develop osteoarthritis at a young age.
- Finger Deformities: Fingers can have "swan-neck" or "Boutonniere" deformities.
- Muscle and Tendon Issues: Tendons or muscles might tear.
- Spine Problems: The spine can curve (scoliosis) or have a hump (kyphosis).
- Pain: Muscle pain (myalgia) and joint pain (arthralgia) can be severe.
- Walking Delays: Babies might walk later than usual.
-

Individual with EDS showing hypermobile fingers, including the "swan-neck" malformation on the 2nd–5th digits, and a hypermobile thumb
-

Individual with EDS displaying hypermobile metacarpophalangeal joints
-
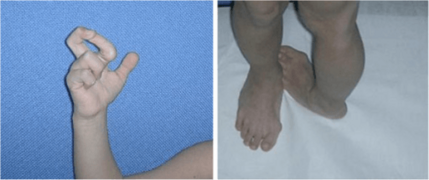
Severe joint hypermobility in a girl with EDS arthrochalasia type



Skin
- Stretchy or Soft Skin: Skin can be very stretchy or feel like velvet.
- Fragile Skin: It tears and bruises easily.
- Unusual Scars: Scars might look thin like cigarette paper.
- Excess Skin Folds: Some types have extra skin folds, especially on the eyelids.
- Thin Skin: In vascular EDS, skin can be thin and see-through.
- Saggy Skin: In dermatosparaxis EDS, skin can be extremely saggy.
-
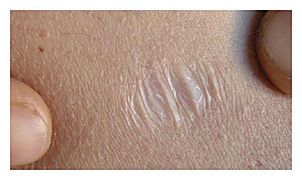
Atrophic scar in a case of EDS
-
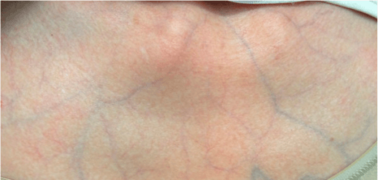
Translucent skin in vascular EDS
-
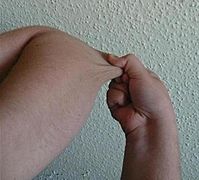
Individual with EDS displaying skin hyperelasticity
-
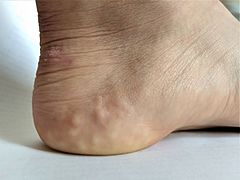
Piezogenic papules on the heel of an individual with hypermobile EDS
-
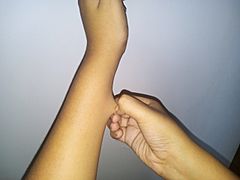
Skin hyperelasticity in the wrist

.png)



Heart and Blood Vessels
- Artery Problems: Arteries can rupture (break open).
- Heart Valve Issues: Heart valves, like the mitral valve, might not work correctly. This can increase the risk of infection during surgery.
- Aorta Problems: The main artery from the heart (aorta) can widen or rupture.
- Blood Pressure Issues: Problems with how the body controls blood pressure.
- Varicose Veins: Swollen, twisted veins can appear.
Other Body Systems
- Digestive Problems: Issues like acid reflux or slow digestion.
- Nerve Problems: Conditions like carpal tunnel syndrome or numbness.
- Dental Issues: Gum disease and weak tooth enamel.
- Eye Problems: Nearsightedness, retinal tears, or blue whites of the eyes.
- Bone Weakness: Bones can be less dense (osteoporosis or osteopenia).
- Learning Differences: Some evidence suggests EDS might be linked to conditions like attention deficit hyperactivity disorder (ADHD) or autism spectrum conditions.
- Pain: The pain from EDS can range from mild to very severe.
-
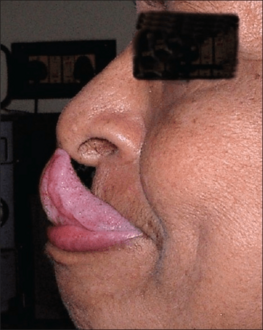
Gorlin's sign in a case of EDS

What Causes EDS?
EDS is caused by changes, or variations, in certain genes. These genes give instructions for making collagen or other proteins that work with collagen. Collagen is like the "scaffolding" that gives strength and structure to connective tissue. When there's a problem with collagen, the connective tissue in the skin, bones, blood vessels, and organs can become weak.
-
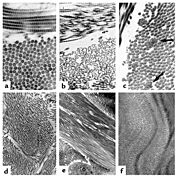
The collagen fibril and EDS: (a) Normal collagen fibrils are of uniform size and spacing. Fibrils from a person with dermatosparaxis (b) show dramatic alterations in fibril morphology with severe effects on the tensile strength of connective tissues. A person with classical EDS (c) shows composite fibrils. Fibrils from a TNX-deficient person (d) are uniform in size and no composite fibrils are seen. TNX-null (e) fibrils are less densely packed and not as well aligned to neighboring fibrils.

Most types of EDS are inherited in an autosomal dominant way. This means a person only needs one changed copy of the gene to have the disorder. A few types are inherited in an autosomal recessive way, meaning both copies of the gene must be changed. Sometimes, the gene change happens on its own during early development and is not inherited from parents.
How is EDS Diagnosed?
Doctors diagnose EDS by looking at a person's medical history and observing their symptoms. They often use a system called the Beighton criteria to check how flexible a person's joints are.
Genetic testing and other lab studies can help identify affected people. However, these tests can't confirm every case, especially if the specific gene change hasn't been discovered yet. So, a doctor's careful evaluation is still very important.
It's common for EDS to be mistaken for other conditions at first. For example, cutis laxa also causes loose skin, but in EDS, the skin stretches and then snaps back. Marfan syndrome also causes very flexible joints and similar heart problems. A correct diagnosis is important to get the right care.
How is EDS Managed?
There is no cure for Ehlers–Danlos syndromes, so treatment focuses on managing symptoms and supporting the body.
- Monitoring: Doctors closely watch the heart and blood vessels.
- Therapy: Physical therapy and occupational therapy can help strengthen muscles and teach people how to protect their joints.
- Support Tools: Braces, casts, or wheelchairs can help stabilize joints and prevent injuries.
- Activity Avoidance: People should avoid activities that might cause their joints to lock or overextend.
- Medication: Medicines can help reduce pain or manage heart, digestive, or other related conditions.
- Special Precautions: Medical staff often take extra care with EDS patients due to the risk of complications. For example, in vascular EDS, chest or abdominal pain is treated as an emergency.
It's important for children with EDS to learn about their condition. They should understand why they might need to avoid contact sports or other very physical activities. They should also be taught not to show off their flexible joints, as this can cause early damage. Emotional support and joining support groups can be very helpful for people and their families.
Managing Pain
Dealing with ongoing pain in EDS often requires a team of doctors and therapists. Pain can be caused by tissue injury or by abnormal nerve signals.
- Exercise: Physiotherapy with gentle exercises can help strengthen core muscles and stabilize joints. Stretching should be slow and gentle to avoid dislocations.
- Therapy: Cognitive behavioural therapy can help people cope with severe, ongoing pain.
- Medications:
* NSAIDs (like ibuprofen) can help with pain from inflammation. * Acetaminophen (like Tylenol) can be used to avoid bleeding risks. * Lidocaine can be applied to painful areas or injected for muscle pain. * For nerve pain, doctors might prescribe certain antidepressants or anticonvulsants.
Managing Dislocations
When a joint dislocates or partly dislocates, muscles can spasm, increasing pain.
- Support: A sling can hold the joint in place and help it relax.
- Comfort: Heat, gentle massage, and mental distractions can help reduce pain and promote relaxation.
- Avoid Casts: Orthopedic casts are often not advised if muscles are still spasming.
Surgery
Sometimes, surgery is needed for unstable joints that cause pain or dislocations. Common surgeries include cleaning out a joint, replacing tendons, or repairing joint capsules.
While surgery can help, it doesn't always guarantee a perfect result. Doctors often prefer non-surgical treatments first. Surgery for people with EDS has extra risks:
- Weak Tissues: Tissues are weaker, making surgery more challenging.
- Fragile Blood Vessels: Blood vessels can be fragile, causing problems during surgery.
- Slow Healing: Wounds often heal slowly or not completely.
It's important to find a surgeon who knows a lot about EDS. People with EDS might also need special care with anesthesia because they can be resistant to some local anesthetics.
What is the Outlook for People with EDS?
The future for people with EDS depends on their specific type. Symptoms can vary greatly, even within the same type. Some people have very few symptoms, while others face major challenges in daily life.
Severe joint instability, ongoing pain, and spinal problems can limit movement. For example, simple tasks like rolling over in bed might cause a dislocation. Other issues like problems with the nervous system or heart can also affect a person's health and quality of life.
Most people with EDS have a normal lifespan. However, those with fragile blood vessels have a higher risk of serious complications, like a sudden arterial rupture. For people with vascular EDS, the average life expectancy is around 48 years.
Possible Complications
- Blood Vessel Issues: Enlarged arteries or tears in artery linings.
- Digestive Issues: A high risk of the colon rupturing in vascular EDS.
- Pregnancy Risks: Increased pain, bleeding, or uterine rupture during pregnancy.
- Hearing Loss: Can occur in some types.
- Eye Problems: Retinal tears, cataracts, or dry eyes.
How Common is EDS?
Ehlers–Danlos syndromes are estimated to affect about 1 in 5,000 births worldwide. Doctors are getting better at diagnosing EDS, so the true number might be even higher.
The hypermobile type of EDS is the most common. Other types are very rare. For example, only a handful of children with dermatosparaxis EDS have been reported globally.
Some types of EDS are more common in certain groups, like Ashkenazi Jews.
History of EDS
Before 1997, there were 10 known types of EDS. Then, the classification system was updated to six main types with clearer names. Doctors now recognize that even more types exist, some only found in single families.
For most types, specific gene changes have been identified, which helps with genetic testing. However, if a genetic test is negative, it doesn't always rule out EDS. This is because not all gene changes have been discovered yet. So, a doctor's clinical evaluation is still very important.
EDS in Society and Culture
EDS might have played a role in the amazing skill of violinist Niccolò Paganini. He could play notes that were very far apart, possibly because of his flexible fingers.
Many sideshow performers in the past had EDS. They were known as the Elastic Skin Man or the India Rubber Man. Today, some people with EDS hold world records for their flexibility. For example, contortionist Daniel Browning Smith and performer Gary "Stretch" Turner are known for their incredible flexibility and stretchy skin.
Famous People with EDS
- Actress Cherylee Houston (uses a wheelchair)
- Drag queen Yvie Oddly
- Actress and activist Jameela Jamil
- Writer and actress Lena Dunham
- Singer Sia
- Singer Halsey
- YouTuber Annie Elainey
- Miss America 2020 Camille Schrier
- Singer-songwriter Mandy Harvey
EDS in Animals
Ehlers–Danlos-like syndromes have been found in Himalayan cats, some domestic shorthair cats, and certain breeds of cattle. It also appears sometimes in domestic dogs. The treatment and outlook for animals are similar to humans. Animals with EDS should not be used for breeding, as the condition can be passed on.
- Animal EDS
-
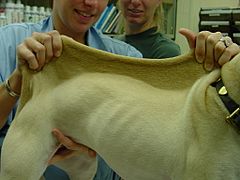
EDS in a dog
-

Same dog with EDS
-
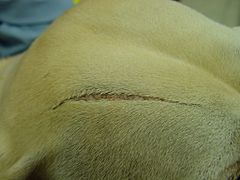
EDS in the same dog showing an atrophic scar



A similar condition called Degenerative suspensory ligament desmitis is seen in many breeds of horses. It was once thought to be only in older horses, but it's now known to affect horses of all ages.
See also
 In Spanish: Síndromes de Ehlers-Danlos para niños
In Spanish: Síndromes de Ehlers-Danlos para niños
- Hypermobility spectrum disorder
