
Distillation facts for kids
Distillation is a clever chemical process. It helps us separate different liquids from each other. Imagine you have a mix of liquids, like water and alcohol. They have different boiling points. This means one turns into a gas (vapor) at a lower temperature than the other.
To separate them, you heat the mixture. The liquid with the lower boiling point turns into a vapor first. This vapor then goes into a special cooling tube called a condenser. The condenser cools the vapor, turning it back into a liquid. This new, separated liquid is called the distillate. What's left behind in the original container is called the "residue."
Sometimes, a special tool called a fractionating column is used. It helps make the separation even better. Distillation is used in many industries. For example, oil refineries use it to clean up crude oil. This makes crude oil useful for many things, like gasoline.
People have used distillation for a long time. It was used to make alcohol for drinks. Distillation can be done anywhere, even at home or in a lab. But in most places, you need a license to distil alcohol. Illegally distilled alcoholic drinks are sometimes called moonshine.
Sometimes, cleaning salty water is called "distillation." But this is a bit different. Water distillation separates a liquid (water) from solids (salts). Alcohol distillation separates two liquids (alcohol and water).
Contents
History of Distillation

People in ancient India knew about distillation. They used clay pots to distil things thousands of years ago. But these early stills could only make very weak liquids. They didn't have good ways to collect the vapors.
Around the 1st century, alchemists in Egypt also knew about distillation. They used it to make Distilled water. Later, in the 3rd century, Zosimus of Panopolis worked on distilling other liquids. In China, distillation of drinks might have started around the 12th or 13th centuries.
A clear record of alcohol distillation comes from the Arab chemist Al-Kindi in 9th-century Iraq. This knowledge then spread to Italy in the 12th century. A special type called Fractional distillation was created by Tadeo Alderotti in the 13th century.
In 1500, a German alchemist named Hieronymus Braunschweig wrote the first book just about distillation. It was called Liber de arte destillandi. Later, in 1651, John French wrote an important English book on distillation. It even showed diagrams of large-scale distillation.





As alchemy became the science of chemistry, new tools were used. These included retorts and alembics. These were glass containers with long, angled necks. The necks acted as simple coolers to turn the vapor back into liquid. Later, copper alembics were invented. These often had a cooling system using cold water. This made the process of collecting alcohol much better. These were called pot stills.
Today, modern methods are often more efficient. But pot stills are still used to make special alcohols. These include cognac, Scotch whisky, Irish whiskey, tequila, and some vodkas. Small pot stills are also used at home to make flower water or essential oils.
Early distillation was a batch process. This means you did one separation at a time. To make things purer, you had to distil the liquid again and again. Sometimes, chemists would do hundreds of distillations to get a very pure substance.
In the early 1800s, new ideas like pre-heating and reflux were developed. In 1822, Anthony Perrier made one of the first continuous stills. This meant the process could run without stopping. Later, Aeneas Coffey improved this design in 1830. His continuous still is like the ancestor of modern industrial distillation units.
By the end of the 1800s, chemical engineering became a science. This allowed for more scientific ways to design distillation processes. The growing petroleum industry in the early 1900s pushed for even better design methods. Today, powerful computers help design distillation columns.
How Distillation is Used
Distillation is used in four main ways:
- In a laboratory (small scale).
- In industry (large scale).
- To get oils from herbs for perfumes and medicines.
- In food processing.
The last two are different. For drinks and herbs, distillation isn't just about making things pure. It's also about moving all the good smells and flavors from the plant into the liquid.
The main difference between lab and industrial distillation is how they run. Lab distillation is often done in "batches." This means you put in a set amount of mixture, separate it, and then start over. In batch distillation, the liquids change as you distil them. The first liquid collected is usually the purest.
In "continuous distillation," the process never stops. New mixture is always added, and separated liquids are always removed. This helps keep the separation process very controlled.
Simple Distillation Basics
When you heat a liquid, it eventually reaches its boiling point. This is the temperature where the liquid's vapor pressure matches the pressure around it. At this point, bubbles form, and the liquid turns into a gas.
It's a common idea that in a mixture, each liquid boils separately. But this isn't quite true. When a mixture boils, all the liquids in it turn into vapor at the same time. However, the lighter liquids (those with lower boiling points) will make up a larger part of the vapor. Heavier liquids will also evaporate, but less of them.
This means that a mixture doesn't have just one boiling point. Instead, its boiling point changes as the lighter liquids boil away. This is why distillation works. The mixture's makeup changes, and so does its boiling point.
Sometimes, liquids don't behave perfectly. For example, alcohol and water can form an "azeotrope." This is a special mixture that boils at a single temperature, like a pure liquid. It can be harder to separate these mixtures completely by simple distillation.
You can't make a mixture 100% pure using only distillation. If you need super-pure products, other separation methods are needed. But for many uses, distillation makes things pure enough.
Batch Distillation

Imagine you have a mixture of two liquids, A and B. Liquid A boils at a lower temperature than B. In a batch distillation setup, you heat this mixture. The vapor that forms above the liquid will have more of liquid A in it. This is because A boils more easily.
This vapor then goes through the condenser and becomes the distillate. As liquid A boils away, the liquid left in the pot becomes richer in liquid B. This causes the boiling point of the mixture to rise. So, the temperature of the vapor also rises. This means the distillate you collect will slowly change, getting more of liquid B over time.
If the boiling points of liquids A and B are very different, the first part of the distillate will be mostly A. Then, as A boils off, the liquid left will be mostly B.
Continuous Distillation
Continuous distillation is a process that runs all the time. A liquid mixture is constantly fed into the system. At the same time, the separated liquids are continuously removed. This process makes at least two output liquids. One is the distillate, which is the lighter liquid that boiled and condensed. The other is the "bottoms" or residue, which is the heavier liquid left behind.
In continuous distillation, the amounts of each liquid stay constant over time. This is different from batch distillation, where they change. Continuous distillation can run for a very long time. To control how pure the liquids are, engineers adjust things like the "reflux ratio." This is how much condensed liquid is sent back into the column. More reflux helps make a better separation.
Laboratory Distillation

In labs, distillation is almost always done in batches. The equipment used is called a still. It has a reboiler or pot to heat the mixture. It also has a condenser to cool the vapor back into a liquid. And finally, a receiver collects the purified liquid, called the distillate. There are several ways to do distillation in a lab.
Simple Distillation
In simple distillation, the vapor goes straight into the condenser. The liquid collected (distillate) isn't perfectly pure. Its makeup is the same as the vapor at that temperature.
Simple distillation works best when the liquids have very different boiling points. A good rule of thumb is a difference of at least 25°C. It's also great for separating liquids from solids or oils that don't boil. In these cases, the liquids separate well enough for most uses.
Fractional Distillation
Often, the boiling points of liquids in a mixture are too close for simple distillation. For these cases, fractional distillation is used. This method separates liquids by doing many boiling and condensing cycles. This happens inside a special tube called a fractionating column. This process is also called rectification.
When the mixture is heated, its vapors rise into the fractionating column. As the vapor goes up, it cools and condenses on the column's walls. This liquid then gets heated again by the rising hot vapors and turns back into vapor. Each time this boiling and condensing happens, the vapor becomes purer in the liquid that boils at a lower temperature. Each cycle is like a "theoretical plate." More theoretical plates mean a better separation.
Steam Distillation
Steam distillation is used for liquids that are sensitive to heat. Instead of directly heating the mixture, steam is bubbled through it. The steam's temperature is easy to control. This allows heat to be transferred well without making the mixture too hot. Some of the target liquid will turn into vapor along with the steam. This vapor mixture is then cooled and condensed. Usually, you get a layer of oil and a layer of water.
Steam distillation of herbs and flowers can make two products. One is an essential oil, which is used in perfumes. The other is a watery herbal distillate, used in aromatherapy and food processing.


Vacuum Distillation
Some liquids have very high boiling points. Instead of heating them to extreme temperatures, it's often better to lower the pressure around them. When the pressure is lowered enough, the liquid will boil at a much lower temperature. This method is called vacuum distillation. It's often used in labs with a rotary evaporator.
Vacuum distillation is also very useful for liquids that would break down if heated to their normal boiling point. By lowering the pressure, they can be distilled without being destroyed.
Industrial Distillation

Large-scale industrial distillation is used in many places. This includes petroleum refineries, chemical plants, and natural gas processing plants. They use continuous distillation methods.
Industrial distillation usually happens in huge, tall towers. These are called distillation towers or distillation columns. They can be very wide and very tall. When a mixture like crude oil is put into these towers, different liquids (called fractions) come out at different levels. The "lightest" liquids (lowest boiling point) come out the top. The "heaviest" liquids (highest boiling point) come out the bottom. These are often called the bottoms.
Industrial towers use something called reflux. This is when part of the condensed liquid from the top of the tower is sent back down. This helps cool the rising vapors and makes the separation much better. The more reflux, the better the separation.
These towers are also used in cryogenic air separation. This process makes liquid oxygen, liquid nitrogen, and pure argon. Distillation of chlorosilanes helps make very pure silicon for semiconductors.
Designing and running these towers depends on what liquids are being separated. For simple mixtures, engineers use special calculations. For complex mixtures, they use computer simulations. The trays or packing inside the towers help the liquids and vapors mix well. This makes the separation more efficient.
In modern industrial uses, a "packing material" is often used instead of trays. This material helps the liquids and vapors interact. It's good for systems where low pressure is needed, or for smaller towers. This packing can be random pieces or structured sheets of metal. Liquids spread over the surface of the packing, and vapors pass through. This is where the separation happens.

Images for kids
-
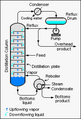
Diagram of a typical industrial distillation tower.
-
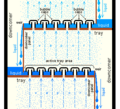
Section of an industrial distillation tower showing detail of trays with bubble caps.


See also
 In Spanish: Destilación para niños
In Spanish: Destilación para niños
