
Metal carbonyl facts for kids


Metal carbonyls are special chemical compounds where transition metals (like iron or nickel) are connected to carbon monoxide (CO) molecules. These compounds are very useful in organic chemistry, which is the study of carbon-based molecules. They act as catalysts, which are substances that speed up chemical reactions without being used up themselves. For example, they help make important chemicals like those used in plastics or detergents.
One famous use is in the Mond process, where nickel tetracarbonyl helps make very pure nickel.
It's important to know that metal carbonyls can be harmful if they touch your skin, are breathed in, or are swallowed. This is partly because they can react with the hemoglobin in your blood, stopping it from carrying oxygen.
Contents
- What are Metal Carbonyls Called?
- How Metal Carbonyls are Built
- How Scientists Study Them
- Where are Metal Carbonyls Found in Nature?
- How Metal Carbonyls are Made
- How Metal Carbonyls React
- Types of Metal Carbonyl Compounds
- What are Metal Carbonyls Used For?
- Similar Compounds
- Safety Concerns
- A Brief History
- Images for kids
What are Metal Carbonyls Called?
The names of metal carbonyls depend on a few things:
- Whether the compound has an electrical charge (positive or negative).
- How many metal atoms are in the center.
- What other groups (called ligands) are attached to the metal.
Sometimes, the carbon monoxide (CO) molecule is attached to just one metal atom. Other times, it acts like a "bridge," connecting two or more metal atoms.
Some metal carbonyls, like nickel tetracarbonyl (Ni(CO)4), only have CO molecules attached. These are called homoleptic. But most metal carbonyls have a mix of CO and other types of molecules attached. These are called heteroleptic.
If a metal carbonyl has only one metal atom, it's called mononuclear. Most neutral mononuclear metal carbonyls have metals with an even atomic number, like chromium, iron, and nickel. If they have more than one metal atom, they are called polynuclear. These often have a direct connection between the metal atoms.
Carbon monoxide can attach to metals in different ways. Usually, only the carbon atom connects to the metal. But sometimes, both the carbon and oxygen atoms connect.

How Metal Carbonyls are Built




Carbon monoxide connects to metals in a special way called "synergistic pi* back-bonding." This means the bond between the metal and carbon has three parts, making it a very strong, almost triple bond.
1. One part is a sigma bond, where electrons from the carbon atom share with the metal. 2. The other two parts are pi bonds, where electrons from the metal move back to the carbon monoxide molecule.
This "back-bonding" makes the bond between the metal and carbon stronger. It also makes the bond inside the carbon monoxide molecule (between carbon and oxygen) a bit weaker than in free carbon monoxide. This is why the distance between the metal and carbon atom is usually very short.
Scientists use a tool called Infrared spectroscopy to study these bonds. It can tell them if a CO molecule is attached to just one metal or if it's bridging between two or more. When CO bridges between two metals, its vibration frequency is lower than when it's attached to just one. If it bridges three metals, the frequency is even lower.
What They Look Like
Most metal carbonyls with one metal atom are colorless or pale yellow liquids or solids. They can catch fire easily and are toxic. Vanadium hexacarbonyl is special because it's a blue-black solid.
Metal carbonyls with two or more metal atoms often have deeper colors. For example, Triiron dodecacarbonyl (Fe3(CO)12) forms deep green crystals. Many of these solid compounds can turn into a gas when heated in a vacuum, but sometimes they break down during this process.
Metal carbonyls can dissolve in many organic liquids like benzene or acetone. Some charged metal carbonyls can even dissolve in water.
How Scientists Study Them

Besides taking "pictures" with X-ray crystallography, scientists use two main tools to study metal carbonyls:
IR spectroscopy is like taking a quick snapshot of the molecule, showing how the atoms are vibrating. NMR spectroscopy is like taking a longer video, showing how the atoms move and swap places over time.
For example, dicobalt octacarbonyl (Co2(CO)8) looks complicated with IR, showing many different ways the CO groups are arranged. But with NMR, it looks simple, showing only one signal. This means the CO groups are moving around and swapping positions very quickly, so NMR sees an average picture.

Iron pentacarbonyl also shows only one signal in 13C NMR because its CO groups quickly swap places in a process called Berry pseudorotation.
Infrared Spectra (IR)

IR spectroscopy is a very important tool. The C–O bond in carbon monoxide gas vibrates at a specific energy (2143 cm−1). In metal carbonyls, this vibration energy changes. It tells scientists how strong the bond is between the carbon and oxygen, and how much "back-bonding" is happening between the metal and carbon.
The more electrons the metal gives back to the CO (stronger back-bonding), the weaker the C–O bond becomes, and the lower its vibration energy will be.
Scientists can use math (called group theory) to predict how many different IR signals a metal carbonyl will show. For example, Cr(CO)6 is shaped like an octahedron and only shows one main IR signal for its CO groups. Compounds with less perfect shapes will show more signals.
| Compound | CO Vibration (cm−1) | 13C NMR shift (ppm) | Metal-CO distance (pm) | C-O distance (pm) | |
|---|---|---|---|---|---|
| CO | 2143 | 181 | |||
| Ti(CO)2− 6 |
1748 | 245 | 204 | 116 | |
| V(CO)− 6 |
1859 | (paramagnetic) | 200, 193 | 113 | |
| Cr(CO)6 | 2000 | 212 | 191 | 114 | |
| Mn(CO)+ 6 |
2100 | ||||
| Fe(CO)2+ 6 |
2204 | 191 | 112 | ||
| Fe(CO)5 | 2022, 2000 | 209 | 180 | 112 | |
| Ru(CO)5 | 2038, 2002 | ||||
| Ni(CO)4 | 181 | 113 |
| Carbonyl | CO Vibration, single (cm−1) | CO Vibration, bridging 2 (cm−1) | CO Vibration, bridging 3 (cm−1) |
|---|---|---|---|
| Rh2(CO)8 | 2060, 2084 | 1846, 1862 | |
| Rh4(CO)12 | 2044, 2070, 2074 | 1886 | |
| Rh6(CO)16 | 2045, 2075 | 1819 |
Nuclear Magnetic Resonance (NMR)
Carbon-13 NMR spectroscopy is another common way to study metal carbonyls. To make it easier to see, scientists often use CO molecules that have a special type of carbon atom (13C).
CO groups attached to just one metal usually show up in a certain range (150 to 220 ppm). CO groups that bridge between metals show up in a different range (230 to 280 ppm). The signals also change depending on the metal atom.
NMR can also show if molecules are "fluxional," meaning their parts are constantly moving and swapping positions.
Mass Spectrometry
Mass spectrometry helps scientists figure out the structure and what elements are in a metal carbonyl. It works by breaking the molecule into smaller pieces and measuring their weight. For metal carbonyls, the most common thing that happens is that CO molecules break off.
- M(CO)+
n → M(CO)+
n−1 + CO
Scientists can also add a charge to neutral metal carbonyls, which helps them analyze the compounds more easily.
Where are Metal Carbonyls Found in Nature?

Believe it or not, metal carbonyls have been found in space! Scientists have detected vibrations of iron carbonyls in the interstellar dust clouds near the center of our Milky Way galaxy. They've also found iron carbonyl clusters in meteorites.
On Earth, metal carbonyls usually break down quickly because of the oxygen in our air. However, some scientists think that in the early Earth's history, in places without much oxygen (like deep-sea vents), these compounds might have formed. They could have acted as catalysts to create important biological molecules.
Tiny amounts of iron, nickel, and tungsten carbonyls have also been found in gases coming from sewage treatment plants.
Some enzymes in living things, like hydrogenase, contain CO attached to iron. This CO helps the enzyme work by keeping the iron in a low oxidation state, which is important for handling hydrogen. Other enzymes, like carbon monoxide dehydrogenase, also deal with CO.
How Metal Carbonyls are Made
Scientists have many ways to make metal carbonyls, from simple ones with one metal atom to complex clusters with many metal atoms.
Direct Reaction with Carbon Monoxide
Some metal carbonyls, like nickel tetracarbonyl and iron pentacarbonyl, can be made by reacting finely divided metal directly with carbon monoxide:
- Nickel + 4 CO → Ni(CO)4 (at 55 °C and normal pressure)
- Iron + 5 CO → Fe(CO)5 (at 175 °C and high pressure)
Nickel tetracarbonyl forms easily at lower temperatures and pressures. Iron needs higher temperatures and pressures.
Reducing Metal Salts and Oxides
Many metal carbonyls are made by reducing metal salts (like metal chlorides) while carbon monoxide is present. "Reducing" means adding electrons. Different chemicals can be used as reducing agents, such as copper, aluminum, hydrogen, or special metal-containing compounds.
For example, chromium hexacarbonyl can be made from chromium(III) chloride using aluminum as the reducing agent:
- CrCl3 + Al + 6 CO → Cr(CO)6 + AlCl3
Sometimes, carbon monoxide itself can act as the reducing agent. For instance, dirhenium decacarbonyl was first made from rhenium oxide:
- Re2O7 + 17 CO → Re2(CO)10 + 7 CO2
If metal oxides are used, carbon dioxide is formed. If metal chlorides are used, a toxic gas called phosgene can be formed.
Using Light or Heat
Shining light on or heating up simple metal carbonyls can create more complex ones with two or more metal atoms. For example, heating iron pentacarbonyl makes diiron nonacarbonyl:
- 2 Fe(CO)5 → Fe2(CO)9 + CO
If you heat them even more, they will eventually break down into the pure metal and carbon monoxide.
Some metal carbonyls are made by taking CO directly from solvents (liquids used to dissolve other substances) like dimethylformamide.
Swapping Parts (Salt Metathesis)
Another way to make mixed-metal carbonyls is by swapping parts between two different compounds. For example, combining KCo(CO)4 with [Ru(CO)3Cl2]2 can create RuCo2(CO)11.
Making Charged Metal Carbonyls
You can make charged metal carbonyls by adding or removing electrons from neutral ones. For example, reducing dimanganese decacarbonyl with sodium creates a negatively charged carbonyl:
- Mn2(CO)10 + 2 Na → 2 Na[Mn(CO)5]
A famous example is disodium tetracarbonylferrate (Na2Fe(CO)4), also known as "Collman's reagent," which is very useful in making organic compounds.
Positively charged metal carbonyls can also be made, for example, by reacting carbonyl halides with certain acids.
How Metal Carbonyls React
Metal carbonyls are important starting materials for making many other organometallic compounds. They can react in several ways:
- Swapping CO for other molecules.
- Gaining or losing electrons.
- Reactions happening directly on the CO molecule.
Swapping CO Molecules
You can replace CO molecules with other types of molecules (called ligands) by heating the compound or shining light on it. Many different molecules can be used, like phosphines or cyanides.
For compounds with 18 electrons (a common stable number for these types of molecules), the CO usually leaves first, creating a temporary compound with 16 electrons. Then, the new molecule attaches.
- M(CO)n → M(CO)n−1 + CO
- M(CO)n−1 + L → M(CO)n−1L
It takes a certain amount of energy to make the CO leave. For example, it's 105 kJ/mol for nickel tetracarbonyl.
Reduction Reactions
Metal carbonyls can react with reducing agents (substances that give electrons) like metallic sodium to form negatively charged carbonyls.
- Fe(CO)5 + 2 Na → Na2[Fe(CO)4] + CO
Reactions at the CO Molecule
The carbon atom in the CO molecule can be attacked by certain chemicals called nucleophiles. For example, a hydroxide ion (OH−) can attack a CO molecule, leading to the release of carbon dioxide and the formation of new compounds. This is known as the "Hieber base reaction."
- Fe(CO)5 + NaOH → Na[Fe(CO)4CO2H]
Other chemicals can also add to the CO molecule, creating different types of compounds.

Reactions with Electrophiles
Even though metal carbonyls usually have metals in low oxidation states, they don't react much with many electrophiles (chemicals that like electrons). However, they do react with halogens (like chlorine or bromine). For example, iron pentacarbonyl reacts with halogens to form iron carbonyl halides:
- Fe(CO)5 + X2 → Fe(CO)4X2 + CO
Halogens can also break the bonds between metal atoms in polynuclear carbonyls.
- Mn2(CO)10 + Cl2 → 2 Mn(CO)5Cl
Types of Metal Carbonyl Compounds
Most metal carbonyls have a mix of CO and other molecules attached. Some common examples include Vaska's complex and methylcyclopentadienyl manganese tricarbonyl. Many of the basic metal carbonyls (those with only metal and CO) are available to buy. Scientists often use a rule called the 18-electron rule to predict what these compounds will look like.
Neutral Metal Carbonyls
Here are some examples of neutral metal carbonyls:
- Group 6 elements (like chromium, molybdenum, tungsten) form hexacarbonyls, such as Cr(CO)6.
- Group 7 elements (like manganese, technetium, rhenium) form pentacarbonyls that link up in pairs, such as Mn2(CO)10.
- Group 8 elements (like iron, ruthenium, osmium) form pentacarbonyls, such as Fe(CO)5. Some of these are not very stable and can form larger clusters like Ru3(CO)12.
- Group 9 elements (like cobalt, rhodium, iridium) often form tetracarbonyls that link up in pairs, like Co2(CO)8. They can also form larger clusters like Co4(CO)12.
- Group 10 elements (like nickel) form tetracarbonyls, such as Ni(CO)4.
Negatively Charged Metal Carbonyls
These are metal carbonyls that have a negative electrical charge.
- Group 4 elements can form dianions (compounds with a 2- charge), like [Ti(CO)6]2−.
- Group 5 elements can form monoanions (compounds with a 1- charge), like [V(CO)6]−.
- Group 8 elements can form dianions, like [Fe(CO)4]2−.
- Group 9 elements can form monoanions, with [Co(CO)4]− being the most studied.
Many negatively charged metal carbonyls can gain a hydrogen atom to form "metal carbonyl hydrides."
Positively Charged Metal Carbonyls
These are metal carbonyls that have a positive electrical charge.
- Group 7 elements can form monocations (compounds with a 1+ charge), like [Mn(CO)6]+.
- Group 8 elements can form dications (compounds with a 2+ charge), like [Fe(CO)6]2+.
What are Metal Carbonyls Used For?

In Metal Production
Metal carbonyls are used in several industrial processes. One of the first uses was in the Mond process to extract and purify nickel using nickel tetracarbonyl.
Similarly, very pure iron powder, called carbonyl iron, is made by heating iron pentacarbonyl until it breaks down. Carbonyl iron is used in many things, including making pigments, as a dietary supplement, and in materials that absorb radar waves for stealth technology.
As Catalysts
Metal carbonyls are important catalysts in many industrial reactions that involve carbon monoxide.
- In the oxo process (also called hydroformylation), metal carbonyls like dicobalt octacarbonyl help combine an alkene (a type of hydrocarbon), hydrogen gas, and carbon monoxide to make aldehydes. For example, it helps make butyraldehyde from propylene:
* CH3CH=CH2 + H2 + CO → CH3CH2CH2CHO Butyraldehyde is then used to make other chemicals for plastics and detergents. This process is very efficient because almost all the starting materials end up in the final product.
- Another important reaction is hydrocarboxylation, which uses metal carbonyls to make things like acrylic acid.
- Metal carbonyls also help in reactions where acetylene (a gas) is turned into larger ring-shaped molecules.
- In the Monsanto process and Cativa process, rhodium and iridium carbonyl catalysts are used to make acetic acid from methanol, carbon monoxide, and water.
CO-Releasing Molecules (CO-RMs)
Scientists are developing special metal carbonyls called "CO-releasing molecules" (CO-RMs). These are designed to release small, controlled amounts of CO in the body. Why? Because in small amounts, CO can act as a medicine, helping to widen blood vessels and reduce inflammation. Scientists hope these CO-RMs could be used as new drugs.
Similar Compounds
Many other compounds are similar to metal carbonyls but have different types of molecules attached instead of or in addition to CO.
Nitrosyl Complexes
These compounds have nitrosyl (NO) molecules attached to metals. Unlike metal carbonyls, it's rare to find a compound with only NO molecules. NO is even better at "back-bonding" than CO. Examples include CoNO(CO)3.
Thiocarbonyl Complexes
These compounds have CS molecules attached. They are not very common because carbon monosulfide itself is unstable. Scientists have to use special ways to make them, like reacting disodium tetracarbonylferrate with thiophosgene:
- Na2Fe(CO)4 + CSCl2 → Fe(CO)4CS + 2 NaCl
Compounds with CSe and CTe have also been made.
Isocyanide Complexes
Isocyanides are molecules similar to CO that also form many complexes with metals. Common examples include methyl isocyanide.
Safety Concerns
Metal carbonyls are toxic because they can release carbon monoxide, and because the metal itself can be harmful. They are often volatile (turn into gas easily) and can be absorbed through the skin, inhaled, or swallowed.
The most common poisonings have happened with nickel tetracarbonyl and iron pentacarbonyl because they are used in industry. Nickel tetracarbonyl is considered one of the most dangerous poisons if inhaled.
Breathing in nickel tetracarbonyl can cause immediate symptoms like nausea, cough, headache, fever, and dizziness. Later, more serious problems can appear in the lungs, stomach, brain, liver, and kidneys. Recovery from metal carbonyl poisoning can take a long time.
Long-term exposure to low levels of nickel tetracarbonyl can cause problems like trouble sleeping, headaches, and memory loss. It is also believed to cause cancer, but this can take many years to show up.
A Brief History

The first experiments with carbon monoxide and metals were done by Justus von Liebig in 1834. He made a substance he called "Kohlenoxidkalium," but it wasn't a true metal carbonyl.
The first real metal carbonyl compound, dicarbonyldichloroplatinum (Pt(CO)2Cl2), was made by Paul Schützenberger in 1868.

In the 1890s, Ludwig Mond and his team were trying to recover chlorine. During their experiments, they treated nickel with carbon monoxide. They noticed a gas that made a green-yellow flame and formed a nickel mirror when heated. This gas could be cooled into a clear liquid that boiled at 43 °C. This is how Mond discovered the first pure metal carbonyl, nickel tetracarbonyl (Ni(CO)4). Its unusual ability to turn into a gas easily led to the saying that Mond had "given wings to the heavy metals."
The next year, Mond and Marcellin Berthelot independently discovered iron pentacarbonyl. Mond quickly saw the business potential of these compounds and used them in his Mond process.
In 1906, James Dewar and H. O. Jones figured out the structure of diiron nonacarbonyl. After Mond passed away in 1909, the study of metal carbonyls slowed down for a few years.
In 1924, BASF started making iron pentacarbonyl for industrial use, to produce very pure iron called carbonyl iron.
Walter Hieber played a huge role in developing metal carbonyl chemistry starting in 1928. He discovered important reactions, including the "Hieber base reaction," which was the first way to make metal carbonyl hydrides. He published many papers on the topic.

In the 1930s, Walter Reppe, an industrial chemist, discovered many important catalytic processes using metal carbonyls. For example, he found ways to make acids from olefins and carbon monoxide. He also discovered how to turn acetylene into benzene using metal carbonyl catalysts.

Scientists later developed the idea of "isolobal analogy" to help design new metal carbonyl compounds. This idea, for which Roald Hoffmann won a Nobel Prize, compares parts of metal carbonyls to parts of organic molecules. For example, a Mn(CO)5 part is "isolobal" to a methyl radical (CH3•). This means they behave similarly in some ways, even though they are very different.
The economic success of reactions using metal carbonyls as catalysts, like Reppe chemistry, led to a lot more research in this area. Scientists also discovered that metal carbonyl compounds are found in the active sites of three natural enzymes.
Images for kids
-
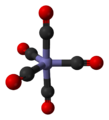
An iron atom with five CO groups attached.
-

A sample of iron pentacarbonyl, which is a liquid that is stable in air.
-
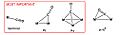
Different ways carbon monoxide can attach to metal atoms.
-

The highest energy electron cloud (HOMO) of CO.
-
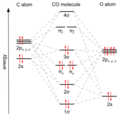
How the electron clouds (orbitals) of carbon monoxide are arranged.
-

The lowest energy empty electron cloud (LUMO) of CO.
-

This diagram shows how electrons move between the metal and carbon monoxide, making a strong bond.
-

Different forms (isomers) of dicobalt octacarbonyl.
-
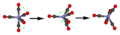
How the CO groups in iron pentacarbonyl quickly swap their positions.
-

This chart shows how many different ways CO groups can vibrate in some common metal carbonyls.
-
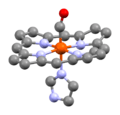
A heme unit in human carboxyhemoglobin, showing the CO molecule attached to the iron atom.
-
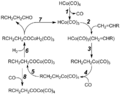
The mechanism of hydroformylation, a reaction catalyzed by metal carbonyls.
-

Hydrocarboxylation of acetylene.
-

Hydrocarboxylation of acetylene with an alcohol.
-
How Fischer carbenes are made using metal carbonyls.
-

Spheres of nickel made using the Mond process.
-

Justus von Liebig (1860).
-

Ludwig Mond, around 1909.
-

The Kaiser Wilhelm Institute for Coal Research (now the Max Planck Institute for Coal Research).
-
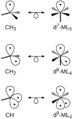
This diagram shows how parts of metal carbonyls can be similar to parts of organic molecules.

